L’enfisema polmonare consiste in una malattia respiratoria cronica, caratterizzata da una vera e propria perdita di tessuto polmonare che viene “consumato” nel tempo, fino a portare a quadri di deficit respiratorio talmente importanti da configurare, in certi casi, un’insufficienza respiratoria.
Giunti a questa fase, purtroppo, le terapie consentono al massimo di ridurre il disagio respiratorio del paziente, minimizzandone i sintomi, primo tra tutti la dispnea (difficoltà respiratoria).
Molti quadri di broncopneumopatia cronica ostruttiva (BPCO) possono presentare, accanto ai classici sintomi conseguenti alla iper-produzione di secreti catarrali (quota bronchitico-cronica della BPCO) e all’ostruzione dei bronchi, anche una quota di dispnea direttamente dipendente dalla dimensione enfisematosa che spesso si ritrova in questa malattia respiratoria.
Tenuto conto che anche un individuo sano, purtroppo, vede ridursi nel tempo la propria capacità funzionale respiratoria, con una perdita progressiva della funzione polmonare stimabile alla spirometria, che segue curve di discesa ben definite (vedi “BPCO, smog e traffico automobilistico urbano: il punto dello pneumologo”), ma con una riduzione della funzionalità respiratoria che difficilmente giunge a quadri tali da compromettere seriamente la vita di relazione e le abitudini della persona, nel caso in cui, invece, il paziente sia esposto nel corso della vita al fumo di sigaretta e allo smog urbano, la progressione di tale deterioramento respiratorio si accentua, fino a danneggiare seriamente la capacità respiratoria del soggetto.
I principali responsabili dell’“invecchiamento” polmonare e della conseguente perdita della sua funzione respiratoria, sono
- le sostanze ossidanti che respiriamo, presenti nel fumo di sigaretta e nell’ambiente, prime tra tutte l’ozono
- le proteasi, cioè quelle particolari proteine (elastasi) prodotte anche da certi globuli bianchi (neutrofili), che digeriscono letteralmente il tessuto elastico interstiziale del polmone.
L’eccesso di ossidanti e di elastasi neutrofila, quindi, danneggia il polmone e, in modo particolare, le pareti dei suoi alveoli, in misura tanto maggiore quanto più elevata è la quantità di queste sostanze presenti nei polmoni.
Visita Pneumologica
Che cos’è (e come viene fatta)?
Scopri tutte le fasi della visita specialistica e come il Dott. Enrico Ballor valuta il paziente per il disturbo che presenta.
Guarda il Video
D’altro canto, esistono molecole naturali protettive ad attività antiossidante (vitamina E, glutatione, ecc,) e anti-proteasica (anti-elastasica), che operano in senso contrario, inattivando l’attività nociva svolta sui tessuti polmonari dalle prime.
Nei soggetti sani, un ben definito equilibrio tra ossidanti/antiossidanti e proteasi/anti-proteasi, fa si che la bilancia tra gli uni e gli altri penda dalla parte delle sostanze ad azione protettiva.
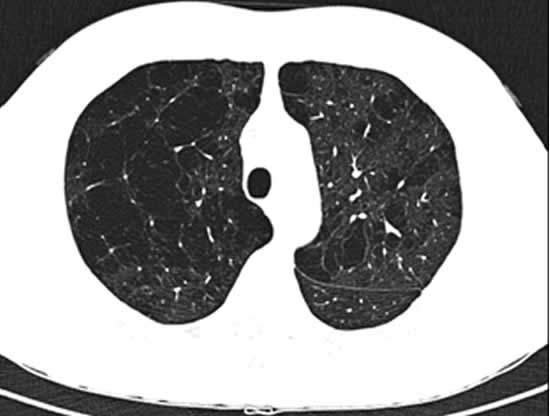
Quando invece, per un qualunque motivo, si viene ad alterare tale rapporto (aumento delle prime e riduzione delle seconde) e le sostanze che svolgono una funzione “aggressiva” sul polmone diventano preponderanti su quelle in grado di proteggerlo, si viene a configurare una situazione caratterizzata dal consumo del tessuto polmonare, che procede poi fino all’enfisema polmonare (vedi “I segni radiologici di enfisema polmonare: il parere dello pneumologo” e l’immagine della TAC del torace qui sotto), con formazione di cavità aeree (bolle di enfisema) formate dalla fusione di più alveoli distrutti, non più in grado di partecipare attivamente allo scambio dei gas respiratori (O2 e CO 2 – vedi “Respiro, polmoni, globuli rossi ed emoglobina: lo pneumologo e la storia della respirazione”).
La principale anti-proteasi naturale (anti-elastasi), proteina ad attività protettiva in grado di contenere e inattivare i danni dell’elastasi neutrofila, è l’α1 -antitripsina (α1-AT), glicoproteina prodotta dal fegato.
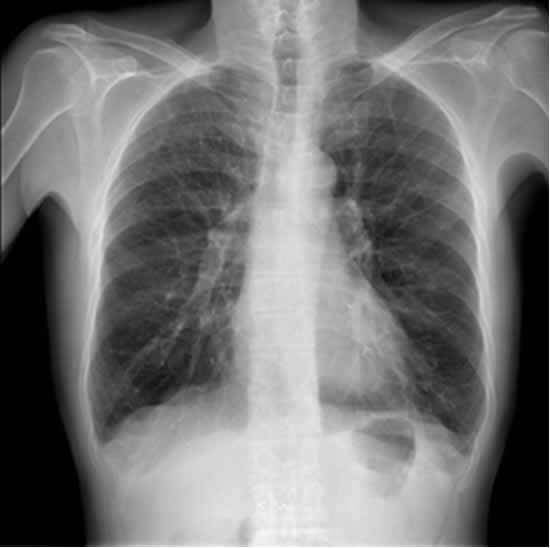
In assenza di essa, il polmone subisce un’importante accelerazione dei processi degenerativi che portano ad un suo progressivo deterioramento funzionale, fino al punto in cui il tessuto polmonare, divenendo estremamente “diradato” e rarefatto, inizia ad apparire più trasparente anche alla radiografia del torace (vedi “Ipertrasparenza enfisematosa, aumentata diafania parenchimale: lo pneumologo spiega il significato”).
Il deficit genetico di tale proteina, peraltro, correla in modo significativo non solamente con la possibilità di provocare enfisema polmonare negli adulti, ma altresì con la possibilità di determinare malattie del fegato nei bimbi.
Per quanto per trattare l’enfisema polmonare si possano prendere in considerazione terapie farmacologiche che tentino di contenere il disagio respiratorio (vedi “Farmaci broncodilatatori nei pazienti con BPCO ed enfisema polmonare”), impiegando farmaci che rappresentano, talora, più delle proposte che non un trattamento risolutivo (vedi “Enfisema polmonare e Aspirina: novità dallo pneumologo”) e potendo, altresì, attivare tutta una serie di misure riabilitative (vedi “Migliorare da subito il proprio modo di respirare” – “Esercizi respiratori per pazienti con BPCO: la ginnastica respiratoria consigliata dallo pneumologo” – “Ginnastica respiratoria, BPCO e Yoga: il parere dello pneumologo”), questa patologia respiratoria rappresenta, tuttavia, uno di quei classici casi in cui la malattia è da curare prima che si manifesti, e non dopo, quando, a danno ormai definito, il paziente rischia di presentare sintomi respiratori anche al minimo sforzo (vedi “”Seduto respiro, ma se mi muovo mi manca subito il fiato”: il punto dello pneumologo”).
Nonostante le terapie chirurgiche ed endoscopiche (con “valvole”) dell’enfisema polmonare si siano proposte tra le possibili opzioni atte a ridurre il disagio respiratorio del paziente, con risultati in molti casi deludenti, specie se posti in relazione con ciò che il paziente si sarebbe atteso da esse, nessuna strategia di cura dell’enfisema si è mai dimostrata in grado di produrre un significativo ripristino del quadro funzionale polmonare.
Di qui, come dicevo prima, la necessità assoluta di procedere in via preventiva, prima che curativa a posteriori.
Come si procede alla diagnosi del deficit di α1-antitripsina?
Nel caso in cui, in presenza di enfisema polmonare documentato radiologicamente e con le prove di funzionalità respiratoria, si sospetti un deficit della proteina come principale causa della malattia, il primo passo da compiere, per confermare la diagnosi, consiste nel dosare l’α1-AT sul siero attraverso un normale prelievo di sangue.
E’ altresì indicato richiedere la PCR, indice biochimico in grado di rilevare un qualsiasi stato infiammatorio dell’organismo, in presenza del quale un eventuale deficit non particolarmente marcato dell’α1-AT (deficit intermedio), potrebbe presentarsi con valori falsamente normali (vedi “Infezioni e infiammazioni bronchiali e polmonari: VES, PCR e procalcitonina”).
Con queste premesse, qualora in presenza di una PCR normale si rilevino, con metodica nefelometrica, valori di α1-AT inferiori a 113 mg/100 ml, è utile procedere con l’indagine diagnostica di conferma della responsabilità genetica del deficit riscontrato.
Si tenga conto del fatto che un livello di α1 -AT superiore a 49 mg/100 ml rappresenta la soglia protettiva al disopra della quale, in ogni caso, pur ridotto rispetto alla normalità, il deficit di α1-AT (DAAT) non è comunque tale da aumentare il rischio di sviluppare un enfisema polmonare da esso dipendente.
L’indagine di conferma del deficit consiste in un semplice test genetico che impiega un prelievo di sangue venoso e un kit diagnostico che consente di conservare il sangue prelevato su appositi dischetti di carta assorbente e di inviarlo, per posta, al laboratorio analisi, ove l’indagine genetica verrà preceduta da una nuova valutazione preliminare di conferma relativa sia all’ α 1-AT, sia alla PCR.
Secondo l’OMS (Organizzazione Mondiale della Sanità), dovrebbero essere sottoposti, almeno una volta nella vita, ad un test quantitativo per il dosaggio dell’α1-AT, tutti i pazienti adulti affetti da BPCO e tutti gli adolescenti che presentino asma bronchiale, specie se in presenza di ostruzione delle vie aeree non completamente reversibile con i broncodilatatori (vedi “I nuovi farmaci per asma e BPCO presentati dallo pneumologo”) .
Questo perché non è tanto il DAAT ad essere una malattia rara, quanto rara è invece la corretta capacità di saperla diagnosticare dopo averci pensato!
Oltre il 90% dei pazienti con DAAT, infatti, non sa di avere il problema, mentre circa il 2% dei pazienti con una diagnosi di BPCO è portatore di DAAT senza saperlo.
Per lo stesso motivo si dovrebbero indagare anche tutti coloro che, pur senza sintomi respiratori, dovessero presentare una persistente ostruzione bronchiale alla spirometria o una malattia epatica che non trovi spiegazione (specie nei bimbi), qualche volta in grado di evolvere fino ai più gravi quadri di cirrosi epatica senza causa nota (cirrosi epatica criptogenetica).
E’, infatti, la presenza dell’allele “Z” che, polimerizzando negli epatociti (cellule del fegato), provoca il danno epatico.
La genetica del deficit di α1-AT sottostà alle regole della trasmissione dei caratteri ereditari previste dalle leggi di Mendel, con genotipi e fenotipi vari dipendenti dal diverso accoppiamento dei diversi “alleli” (normale “M” e anomali “Z” o varianti S e null) nel corredo genetico del paziente.
Da essi dipende la gravità della condizione deficitaria e l’eventuale successiva evoluzione patologica bronco-polmonare ed epatica, strettamente correlata al deficit geneticamente determinato.
Nelle forme di enfisema polmonare nelle quali si dimostri, sul sangue, la presenza di un deficit (specie se grave) di α1-antitripsina (non tutti i casi di enfisema polmonare sono di questo tipo), e se ne stabilisca successivamente, con il test di cui sopra, la definitiva responsabilità genetica, esiste, accanto a tutte le classiche misure volte a ridurre l’esposizione agli inquinanti dell’aria e al fumo di sigaretta (vedi “BPCO e fumo di sigaretta: il parere dello pneumologo” – “Fumo di sigaretta e inquinamento urbano: individuazione precoce dei danni funzionali” – “Bronchite cronica e regole di vita: lo pneumologo e la terapia oltre i farmaci”), la concreta possibilità di trattare il paziente con somministrazioni endovenose settimanali di α1-AT altamente purificata di derivazione umana (derivato del plasma) – (60mg/Kg di peso corporeo, in circa 15 minuti di infusione), ottenendo valori circolanti della proteina deficitaria pari a quelli di un individuo sano, e una riduzione del declino respiratorio pari fino a un terzo del FEV 1 (parametro funzionale che misura l’ostruzione delle vie aeree) rispetto a quello che si sarebbe avuto in assenza di trattamento.
Vantaggio derivante dalla somministrazione di α1-AT per via endovenosa
Il vantaggio derivante dalla somministrazione di α1-AT per via endovenosa, è stato dimostrato da studi internazionali pubblicati, attraverso le immagini inspiratorie ottenute con la TAC del torace ad alta definizione.
In alcuni studi, anzi, sembrerebbe più promettente, sull’efficacia finale complessiva del trattamento, il raddoppio della posologia del farmaco (120mg/Kg di peso corporeo, in circa mezz’ora di infusione), rispetto ai dosaggi oggi ufficialmente autorizzati, in quanto in grado di ottenere livelli di α1-AT circolante più prossimi a quelli degli individui sani.
Naturalmente, sia per la complessità della materia trattata, sia per i costi assai elevati di questo trattamento, in nessun caso il paziente è autorizzato all’autogestione della terapia, che va effettuata, solo dopo accertamento genetico della diagnosi, unicamente da personale qualificato, presso specifici centri specialistici pneumologici di riferimento.
Conclusione
In sintesi, quindi, il deficit di α1-AT (DAAT) consiste in una malattia ereditaria in grado di interferire, talora anche in modo rilevante, con la funzione respiratoria del paziente, con una gravità dei quadri clinici respiratori, talora mortali, direttamente correlata all’entità del deficit genetico della proteina circolante.
Spesso questi pazienti vengono per lungo tempo trattati come “comuni” BPCO o come asmatici “complessi”, con quadri respiratori che stentano a risolversi nonostante la messa in atto di un’adeguata terapia con i farmaci broncodilatatori.
Si tenga conto, inoltre, che in considerazione della natura geneticamente trasmissibile del problema agli altri membri della famiglia, nei casi in cui un paziente presenti una DAAT accertata, risulta utile uno screening famigliare sui parenti, in particolar modo se fumatori.
Visita Pneumologica
Che cos’è (e come viene fatta)?
Scopri tutte le fasi della visita specialistica e come il Dott. Enrico Ballor valuta il paziente per il disturbo che presenta.
Guarda il Video
Vuoi saperne di più?
Faccio il possibile per scrivere i miei articoli informativi nel modo più chiaro, semplice e soprattutto esaustivo che si possa.
Consiglio sempre ai miei lettori di leggere innanzitutto i link di approfondimento che propongo su ciascun articolo, nonché di visitare il portale Medicina del Respiro.

Hai bisogno di un parere medico per il tuo caso?
Non ci sono problemi.
Se credi che il tuo problema abbia carattere di urgenza e debba ricevere una consulenza medica specialistica puoi prenotare da qui una visita privata presso il mio studio.
Hai apprezzato questo articolo?
Realizzare questo sito e i suoi contenuti ha richiesto anni di lavoro.
Non solo: creare continuamente articoli informativi richiede un sacco di tempo, come puoi immaginare.
Non c’è miglior grazie che io possa ricevere da te se non sapere che condividerai questo articolo su Facebook, Twitter o LinkedIn.
A presto!


